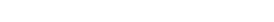İÇİNDEKİLER
- Önsöz
- Giriş
- Biyoteknolojik İlaçlar ve Üretim Süreçleri
- Biyobenzer İlaçlar ve Karşılaştırılabilirlik
- Biyobenzer Ürünlerde Ekstrapolasyon
- Biyobenzer İlaçlarda Değiştirilebilirlik
- Biyoüstünler
- Referans Ürünle Yeterli Karşılaştırma Çalışması Olmayan Biyolojik Ürünler
- Biyolojik ve Biyobenzer İlaçlarda Farmakovijilans, İzlenebilirlik ve Risk Yönetimi
- Yazarlar


PREKLİNİK KARŞILAŞTIRILABİLİRLİK
Preklinik çalışmaların amacı, klinik çalışma öncesi insanda güvenli, etkili başlangıç dozunu bulmak, toksisiteye maruz kalabilecek organları tespit etmek, varsa toksisitenin geri dönüşümlü olup olmadığını belirlemek ve klinikte monitorizasyon için güvenlilik parametrelerini tayin etmektir. Biyobenzer ilaçlar için kısaltılmış preklinik program gereklidir.
Biyolojik ilaçların gelişim sürecinde özellikle biyoteknolojik ilaçların ruhsatlandırılması, tanımlanmış standart preklinik ilaç çalışmalarının olmaması nedeniyle, ilaç otoriteleri arasında uyumsuz kararların çıkması söz konusu olmuştur. Standardizasyon özellikle preklinik çalışmalarda uygun hayvan türü seçimi, immünojenisite çalışmaları, rutin preklinik testler (FK/FD, toksisite…) için ciddi bir gereksinimdir. Standardizasyon ilkeleri her ilaç grubunda farklılıklar göstermekte hatta kimi ilaç gruplarında dosya bazında değerlendirme gerekebilmektedir.
Preklinik çalışmaların standardizasyonu amacıyla öncelikle EMA ilk kılavuzu yayınlamıştır (EMEA/CHMP/BMWP/42832/2005).

Bu kılavuza göre preklinik çalışmaların amacı, klinik çalışma için insanda güvenli, etkili başlangıç dozunu bulmak, toksisiteye maruz kalabilecek organları, varsa toksisitenin geri dönüşümlü olup olmadığını tespit etmek ve klinikte monitorizasyon için güvenlik parametrelerini bulmak olarak tarif edilmiştir (1). Özellikle beş alt başlık tartışılmıştır:
1. Hayvan türü seçimi
2. Çalışma tasarımı
3. İmmünojenisite
4. Üreme ve gelişimsel toksikoloji, genotoksisite
5. Karsinojenisite
EMA yaklaşımı, bu ICH kılavuzunda, tüm ürünler için tek bir kuralın bu alanda uygulanamayacağı esasına dayanmaktadır.
Preklinik çalışmalara genel yaklaşım
Joy A. Cavagnaro / Nature Reviews Drug Discovery 1, 469-475 June 2002’ den, 2. kaynaktan uyarlanmıştır)
Hücre kültüründe (in vitro) yapılan çalışmalar
Biyoteknolojik ilaç adayının öncelikle biyolojik karakterizasyon / aktivasyon çalışmaları in vitro şartlarda gösterilir. İlacın terapötik etkisinin ve etki mekanizmasının anlaşılması amacıyla reseptöre bağlanma çalışmaları (afinite), hücre siklusuna etki, hücre içi yolakların inhibisyonu, DNA tamir mekanizmaları ve apopitozise etkisi araştırılır. İlacın terapötik alanına özgü damarlanma üzerine etki, hücre göçü ve tümör hücresi öldürme gibi spesifik araştırmalarda özellikle in vitro şartlarda hücre kültüründe çalışılır.
Deney hayvanı tür seçimi ve farmakoloji çalışmaları
İki farklı tür, “rodent” ve “non-rodent” seçilir. Bu seçim yapılırken ürünün reseptör / epitop dağılımı mutlaka incelenmelidir. Epitop, antijenin özgüllüğünü belirleyen ve antijenin kendi özgül antikorlarıyla birleşmesini sağlayan kimyasal gruplardır (determinant). Reseptör / epitop dağılımı özellikle “yanıtsız hayvan” türleri için önemlidir. Reseptör veya epitop ekspresyonu yoksa hayvan türü uygunsuzdur. Ekspresyon yapan transjenik hayvanlara gereksinim doğar (2).
Preklinik farmakodinami çalışmaları
Farmakodinami hasta veya normal organizmalar üzerinde ilaçların etkisinin araştırılmasıdır. İlacın etki gücü ve canlı organizma üzerindeki diğer etkiler (in vivo çalışmalarıyla), etki mekanizması, reseptör-ilaç ilişkisi, doz-etki ilişkisi ve ilacın etkisini değiştiren faktörler deney hayvanlarında preklinik çalışmalarda incelenir.
Preklinik farmakokinetik çalışmalar
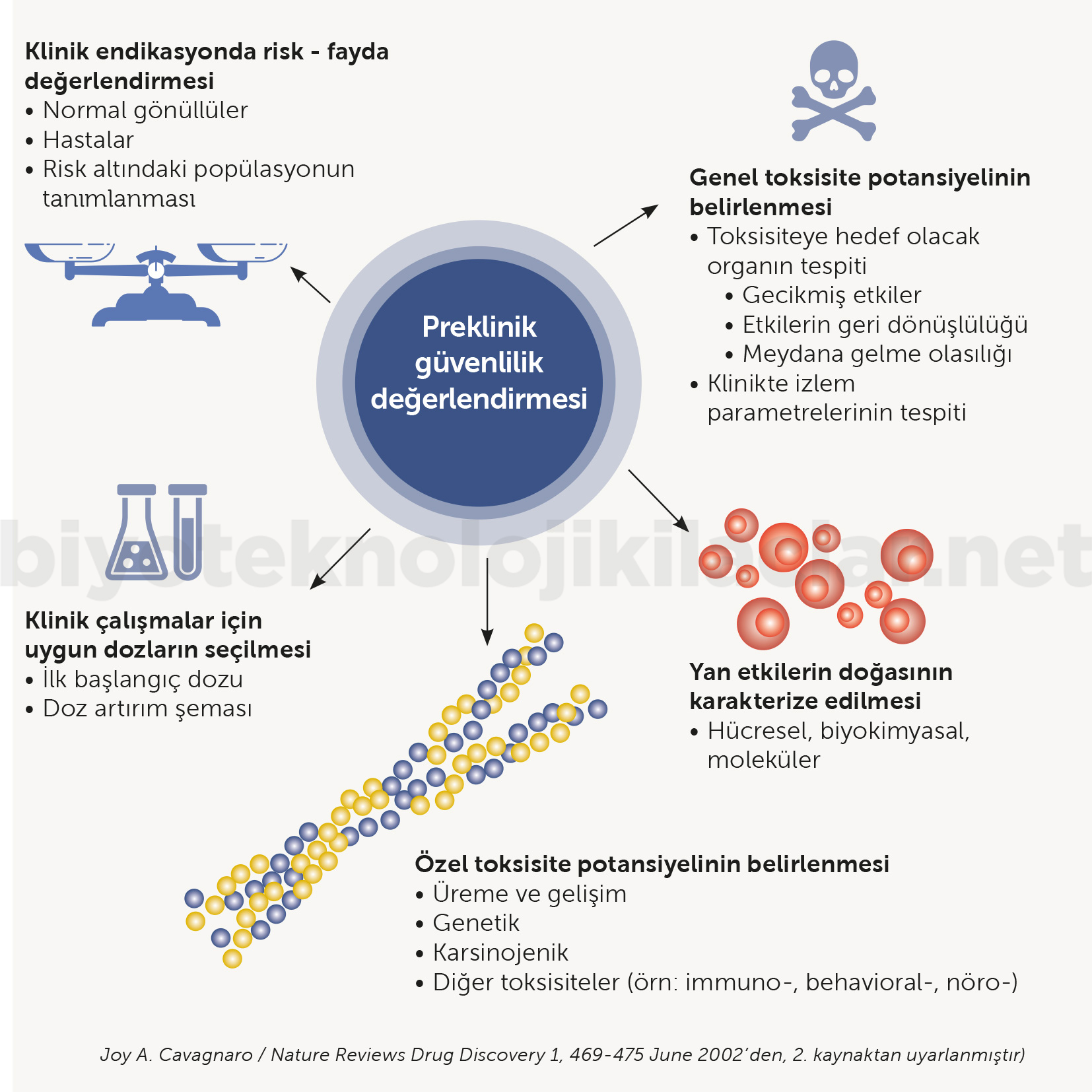
Farmakokinetik çalışmalarda EDMA verileri, maksimum doz tespiti, klerens, ilacın yarılanma ömrü gibi parametreler bulunur. İşaretlenmiş proteinlerin dokuda ölçümüyle ilaç konsantrasyonu tespit edilir.
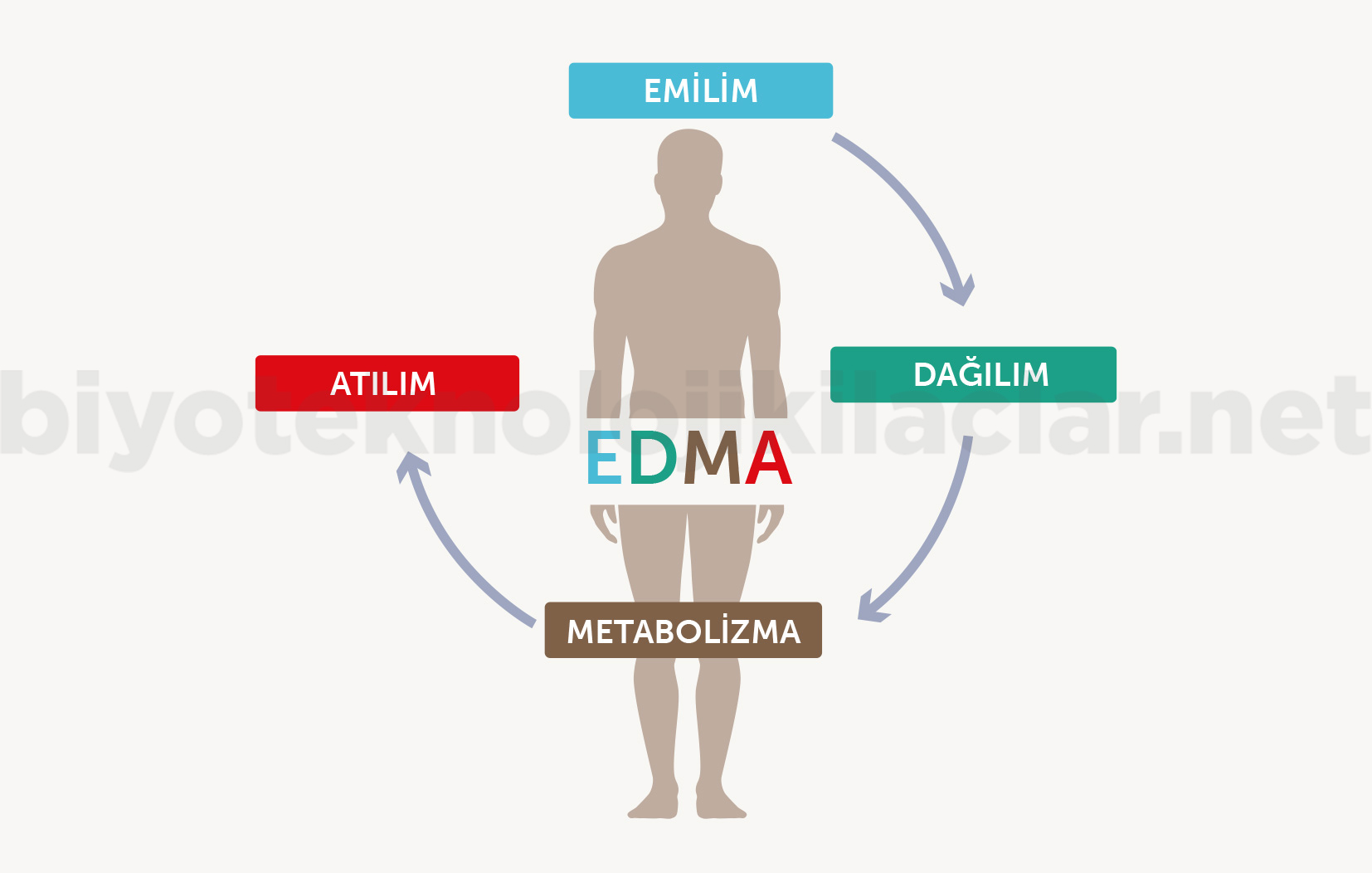
FD / FK çalışmaları sonucunda klinik çalışmaya ön bilgi olarak:
1. Uygulama yolu ve sıklığı
2. Doz seçimi
3. Doz-yanıt ilişkisi
4. Maksimum doz, toksik doz
5. Biyoyararlanım parametreleri elde edilir.
Farmakolojik Güvenlilik, Toksikoloji (“Safety Pharmacology”)
Üretim aşamasında kontaminasyon ve ortamdan uzaklaştırılamayan safsızlıklar biyoteknolojik ilaç advers etkileri açısından son derece önemlidir (3). FD / FK bulgular kullanılarak güvenlilik çalışmaları başlatılır. Çalışmalarda genel protokol:
1. Tek yüksek doz (14 gün takip),
2. Tekrarlı (çoklu) doz (40 hafta takip) şeklindedir.
Subkronik ve kronik değerlendirmede hasarın geçici / kalıcı olması, ilacın dozu, maruziyet süresi ve özellikle toksisitede hedef organlar değerlendirilir.
Güvenlilik çalışmaları için deney hayvanı tür seçimi de önemlidir. Çalışmalar için iki farklı tür belirlenir. Hayvan çalışmaları 3R (replacement, refinement, reduction) kuralı göz önünde bulundurularak tasarlanmalıdır. Eğer ürünün biyolojik aktivitesi çok iyi biliniyorsa bazı durumlarda tek tür kullanılmaktadır. Gene bazen kısa dönem toksisite için iki, uzun dönem toksisite için tek tür kullanılmaktadır.
Birçok ürün, tür ve/veya dokuya özgündür. Standart toksisite testleri bazen işe yaramayabilir. Bu durumda özellikle “yanıtsızlıkta” sadece kardiyovasküler sistem ve solunum fonksiyonları değerlendirilir.
Güvenlilik çalışmaları sırasında deney hayvanlarında takip edilen parametreler çeşitlilik gösterir. Bunlar:
1. Klinik gözlem, davranış takibi (toksisite semptom ve belirtileri için)
2. Vücut ağırlığı ve değişimler
3. Beslenme ve su tüketimi
4. Klinik patoloji verileri (organ hasarı belirteçleri)
Serum enzim, elektrolit, glukoz ve lipid profilleri
Hematolojik inceleme
İdrar analizi
5. Farmakokinetik / Toksikokinetik takip (Toksisite doza ve uygulama şekline bağımlı mı?)
6. Patoloji verileri
İmmünotoksisite değerlendirme çalışmaları
Immünotoksisite (immünoreaktivite) aşırı farmakodinamik yanıt, antikor oluşumu (bazen sağlıklı dokuya) veya immün kompleks oluşumu şeklinde gözlenir. Preklinik çalışmalarda deney hayvanı olarak genelde immün yanıtın en kolay tespit edildiği kobay (“guinea pig”) kullanılır. Klinikte özellikle monoklonal antikorlar ve sitokinler için öncelikli olan anafilaksi değerlendirmesi gözlemsel olarak yapılır. Ayrıca ELISA ile antikor ölçümü gibi yöntemlerle de değerlendirme yapılır. Her ürüne özgü antikor tayin kitleri kullanılmalıdır.
Reprodüktif ve gelişimsel toksisite değerlendirme çalışmaları
Biyolojik ilaç adayının deney hayvanlarında genel üreme performansı, fertilite, peri-postnatal toksisite (gebelik) sürecine etkisi değerlendirilir. Olası embriyotoksisite değerlendirilmesi yapılır.
Genotoksisite değerlendirme çalışmaları
Proteinlerin DNA ile etkileşmesi zor olmakla birlikte, olası mutajenik etki değerlendirilir. Mutajenlerin birikimi kanser sebebi olabilir. Yüksek dozda peptit, protein uygulama DNA hasarına neden olabilir. Sitogenetik ve genotoksisite değerlendirmesi yapılmalıdır.
Karsinojenisite değerlendirme çalışmaları
Yüksek dozlarda kullanılan büyüme faktörleri ve immünosupresifler kansere sebep olabilirler. İn vitro veya in vivo modellerde çalışma ve gözlem yapılır.
Lokal tolerans çalışmaları
Lokal uygulamaya bağlı olası toksisite, enjeksiyon yeri iritasyonu deney hayvanlarında test edilir.
Uyarı
Hayvan çalışmaları klinikte neler olabileceğinin en iyi tahmin yöntemleridir. Bu preklinik modeller mekanizmaların ve olası toksisitelerin incelenmesi açısından faydalıdır. Ancak hiçbir hayvan modeli insanda gözlenen ilaç yanıtına %100 uymaz. Hayvan çalışmaları bazen güvenlilik konusunda aldatıcı olabilir. Unutulmaması gereken bir diğer önemli nokta da bu çalışmaların sağlam hayvanlarda yapılmakta olduğu, ancak ilaçların hasta insana verildiğidir.

Biyobenzer ilaçlarda preklinik çalışmalar:
Biyobenzer ilaçlar orijinal biyoteknolojik ürünlerin (formülasyon) patent süresi dolduktan sonra üretilen benzer versiyonlarıdır. Biyobenzer ilaçlar, orijinal ilaçlarla biyolojik ürün anlamında benzer, fakat özdeş, aynı değildir. Biyoteknolojik ilaçların ekonomiye önemli oranda yük getiriyor olması, sağlık otoritelerini biyobenzer ilaçlara yönlendirmektedir. Avrupa’da orijinal ilaçların patent sürelerinin dolmasından sonra ilk beş biyobenzer ilaç, 2006 ve 2007 yıllarında ruhsat almıştır (4). Biyobenzer ilaçların geliştirilmesiyle dünya genelinde biyolojik ilaçlara olan yüksek ihtiyacın daha uygun maliyet ile karşılanması, rekabetin sağlanması ve araştırma / geliştirmenin teşvik edilmesi hedeflenmektedir. Biyobenzer ilaç geliştirmek için iki önemli faktör söz konusudur:
1. Üretim maliyetinin referans moleküle göre daha düşük olması,
2. Üretim sürecinin referans moleküle göre daha kısa olmasıdır.
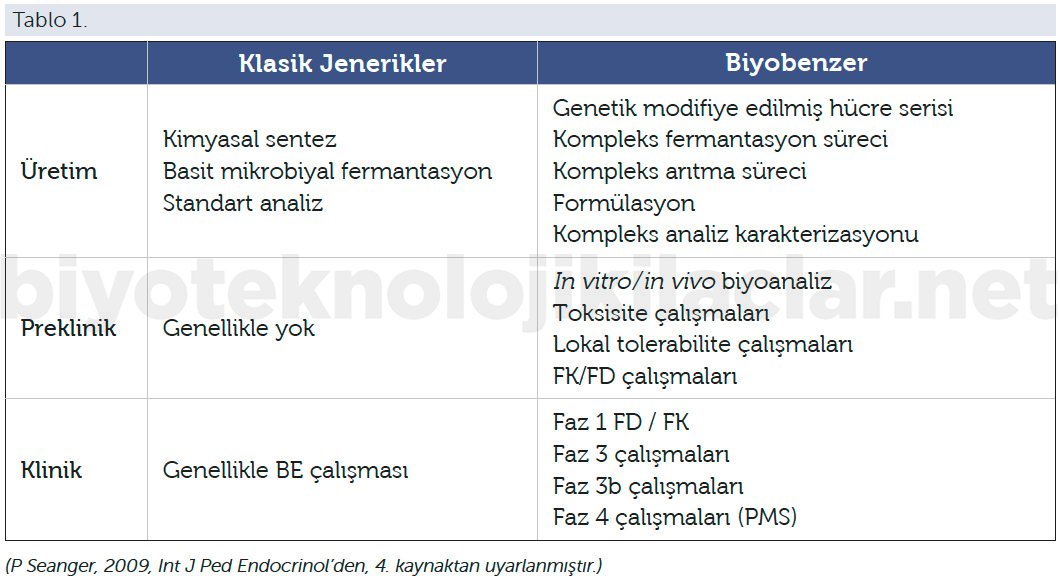
Bu nedenle preklinik ve klinik çalışmalarda ürün bazında farklılıklar görülmekle beraber, genel anlamda referans molekül biyoteknolojik ürünlere göre preklinik değerlendirme açısından daha az, daha sınırlı çalışmalar ve veriler istenir (Tablo 1):
1. Kısaltılmış preklinik program istenir.
2. Subkronik toksisisite çalışması (4 hafta),
3. Lokal tolerans çalışması,
4. Her yeni biyobenzere özgü FK/FD değerleri ve çalışması talep edilir.
KAYNAKLAR