İÇİNDEKİLER
- Önsöz
- Giriş
- Biyoteknolojik İlaçlar ve Üretim Süreçleri
- Biyobenzer İlaçlar ve Karşılaştırılabilirlik
- Biyobenzer Ürünlerde Ekstrapolasyon
- Biyobenzer İlaçlarda Değiştirilebilirlik
- Biyoüstünler
- Referans Ürünle Yeterli Karşılaştırma Çalışması Olmayan Biyolojik Ürünler
- Biyolojik ve Biyobenzer İlaçlarda Farmakovijilans, İzlenebilirlik ve Risk Yönetimi
- Yazarlar


BİYOBENZER İLAÇLAR VE KARŞILAŞTIRILABİLİRLİK
Kalite düzeyinde karşılaştırılabilirlik çalışmaları, etkin madde ve bitmiş ürün için yeterli duyarlılıktaki analitik ve biyoanalitik yöntemlerle gerçekleştirilmelidir.
Biyobenzer ürünlerin referans ilaçlarla kalite açısından karşılaştırılabilirliği aşağıdaki parametreler değerlendirilerek yapılmalıdır (1,2). Kalite verilerini oluşturmak üzere, referans ilaçla karşılaştırılabilirlik çalışmalarında referans ilaç ve biyobenzerin en az 3 farklı serisi kullanılmalıdır (1).
- Biyobenzer ilacın üretim süreci
- Referans ilaçla karşılaştırılabilirlik çalışması
- Referans ilaç seçimi
- Analitik karakterizasyon
- Fizikokimyasal özellikler
- Biyolojik aktivite
- İmmünokimyasal özellikler
- Saflık, safsızlık ve kontaminantlar
- Miktar tayini
- Spesifikasyonlar
1. Biyobenzer İlacın Üretim Süreci
Biyobenzer ilaçların üretim sürecinde Hedef Ürün Kalite Profili (HÜKP) oluşturulmalıdır. Hedef Ürün Kalite Profili (HÜKP) seçilmiş referans üründen sağlanan bilgiler, ulaşılabilir kaynaklar ve referans ürünün kapsamlı karakterizasyonundan elde edilen veriler ile belirlenir. Biyobenzer ilaç geliştirilmesi sürecinde, üretim işlemleri HÜKP esas alınarak yapılır (1-3). Biyobenzer ilacın molekül özellikleri referans ilaçla karşılaştırılabilir olmalıdır. Seçilen ekspresyon sisteminin atipik glikozilasyon paterni veya farklı bir safsızlık profili gibi istenmeyen sonuçlara yol açmaması gereklidir.
Biyobenzer ilaçların üretim süreci bitmiş ürünün özelliklerine etki eden pek çok basamaktan oluşmaktadır ve üretim süreci ürünün kendisini oluşturur.
Üretim kontrollü ve sınıflandırılmış alanlarda cGMP kuralları çerçevesinde gerçekleştirilir (4).
Hücre bankaları kalite değerlendirmesinin başlangıç noktalarıdır. Üretimde kullanılan hücre bankalarının İyi Üretim Uygulamaları (Good Manufacturing Practice- GMP) sertifikaları, hücre bankalarının geliştirilmesi, karakterizasyonu ve klon stabilitesine ait veriler bulunmalıdır. Ana hücre bankası ve çalışma hücre bankalarının karakterizasyonlarının yapılmış olması gereklidir.
Hücre bankalarının karakterizasyonunda aşağıdaki parametreler göz önüne alınır (5).
- Canlılık
- Stabilite
- Beklenmedik virüsler
- Tanıma
- Sterilite
- Retrovirüs
- Saflık
- Mikoplazma
Formülasyon ileri teknoloji dikkate alınarak seçilmelidir. Formülasyon referans ürün ile aynı olmak zorunda değildir (1,6). Formülasyonun stabilite, geçimsizlik (yardımcı maddelerle, seyrelticilerle ve ambalaj materyalleriyle), etkin maddenin bütünlüğü, biyoaktivitesi ve potensi açısından uygunluğu değerlendirilir (7,8).
Biyobenzer ilacın; kalite açısından karşılaştırılabilirliğinin gösterileceği çalışmalar, ticari seriler üzerinde yapılır (5). Referans ilaçtan farklı bir formülasyon ve/veya kap/kapak sistemi seçildiyse (ürünle temas eden herhangi bir materyal varsa) bunun biyobenzer ilacın etkililiği ve güvenliliği üzerinde etkisi olmadığı doğrulanır (9).
Stabilite, ilgili kılavuzlara uygun olarak gerçek zamanlı stabilite çalışmalarıyla ortaya konmalıdır. Etkin madde ve bitmiş ürünün saklama kabındaki stabilitesi değerlendirilmelidır. Parçalanma profilinin değerlendirilmesi için karşılaştırılmalı hızlandırılmış ve stres stabilite çalışmaları yapılmalıdır (1-3,10-12). Stabilite referans ilaçtan ekstrapole edilemez.
Her biyobenzer ilacın gelişim süreci o ürüne özeldir. Ürün geliştirme sırasında, üretim sürecinde değişiklikler yapıldığı zaman (etkin madde ve/veya bitmiş ürün) karşılaştırılabilirlik çalışması yapılmalıdır ve bu çalışmalar biyobenzer karşılaştırılabilirlik çalışmasından ayrı tutulmalıdır.
2. Referans İlaç ile Karşılaştırılabilirlik Çalışması
Referans ilaç seçimi
Referans ilaç, tam dosyayla ruhsatlandırılmış olmalı ve bütün karşılaştırılabilirlik çalışmaları aynı referans ilaçla yapılmalıdır. Referans ilacın ticari ismi, farmasötik formu, formülasyonu, gücü, kaynağı, seri numarası ve son kullanma tarihi, kullanım şekli açıklanmalıdır. Referans standartlar, referans ilaç olarak kullanılamaz (1,2).
Analitik karakterizasyon
Biyobenzer ilacın ve referans ilacın kapsamlı karakterizasyon çalışmaları birbirine paralel olarak, en gelişmiş teknoloji kullanılarak, valide edilmiş yöntemlerle yapılmalıdır. Biyobenzer ilaçların karakterizasyonu etkin madde ve bitmiş ürün düzeyinde yapılmalıdır. Proteinlerin kompleks yapılarından dolayı birden fazla analitik yöntem kullanılması önerilir. Analitik yöntemlerin validasyonları yapılmalı, yöntem standardizasyonu için farmakope standartları kullanılmalıdır. Kantitatif veriler için istatistiksel analiz yapılmalıdır.
Biyobenzer bir ürünün analitik karakterizasyonu için aşağıdaki özelliklerinin tayin edilmesi gereklidir (1,2,11).
- Fizikokimyasal özellikler
- Biyolojik aktivite
- İmmünokimyasal özellikler
- Saflık, safsızlık ve kontaminantlar
- Miktar tayini
1. Fizikokimyasal özellikler
Fizikokimyasal analizlerle etkin maddenin bileşimi, fiziksel ve kimyasal özellikleri, birincil ve kompleks yapıların tayini yapılır. Biyobenzer ilacın hedef amino asit dizisi referans ilaçla aynı olmalıdır. N- ve C- terminal amino asit dizisi, serbest -SH grupları, disülfit köprüleri uygun yöntemlerle karşılaştırılmalıdır. Uygun yöntemlerle post translasyonel modifikasyonların varlığı ve sayısı gösterilmelidir. Karbonhidrat yapıları varsa, genel glikan profili karşılaştırılmalıdır. Her bir biyobenzer ilaca ait üretim süreci sürdürülebilir olmalıdır.
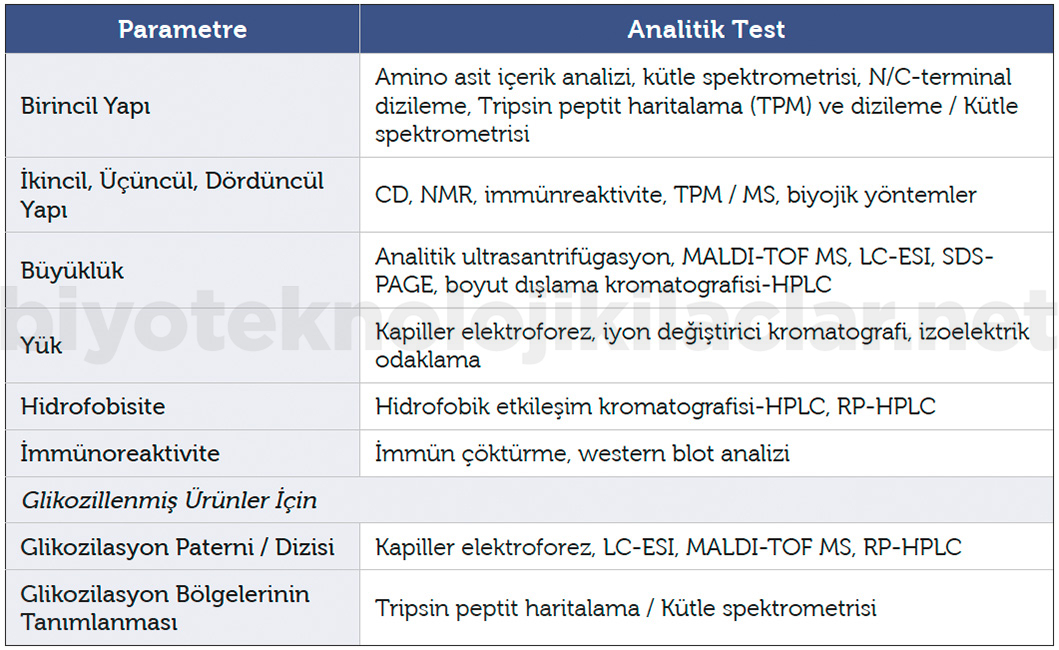
Etkin maddenin bileşimi, fiziksel ve kimyasal özellikleri, büyüklük, yük, hidrofobisite, birincil ve kompleks yapıların tayini için kullanılan pek çok analitik yöntem mevcuttur. Bu yöntemler Tablo 1’de görülmektedir (13-15).
Tablo 1. Etkin madde yapı tayininde kullanılan yöntemler
2. Biyolojik aktivite
Ürünün tanımlanmış biyolojik etkisini gösterebilen kapasitedir. Biyolojik aktivitenin kantitatif ölçümü potens, birimi ünitedir. Biyolojik deneylerin sonuçları ulusal veya uluslararası referans standartlara karşı kalibre edilmiş aktivite ünitesi olarak ifade edilir. Biyolojik aktivite tayin yöntemleri çeşitlidir ve biyolojik aktiviteyi ölçmek için üretici tarafından valide edilmiş ürüne özgü biyolojik yöntemler gereklidir (1,2,11).
Biyolojik aktivite ölçme yöntemleri:
- Hayvan esaslı biyolojik yöntemler: Organizmanın ürüne karşı olan biyolojik yanıtını ölçer.
- Hücre kültürü esaslı biyolojik yöntemler: Hücresel seviyede biyokimyasal ve fizyolojik yanıtın ölçülmesini sağlar.
- Biyokimyasal yöntemler: Enzimatik reaksiyon oranları ve immünolojik etkileşimlerle indüklenen biyolojik yanıtları ölçer.
- Ligand ve reseptör bağlanma deneyleri
3. İmmünokimyasal özellikler
Biyoteknolojik ürün monoklonal antikor (Mab) ise immünolojik fonksiyonları değerlendirilir. Burada Mab’ların Fc kısmının reseptöre bağlanma afinitesi değerlendirilir (FcγR, C1q, FcRn gibi) (1,2,11).
4. Saflık, Safsızlık ve Kontaminantlar
Saflığın tanımlanması analitik yöntemlere bağlıdır ve sonuçlar yöntem bağımlıdır. Saflık spesifik aktiviteyle ifade edilir (biyolojik aktivite ünitesi / mg ürün). Safsızlıklar üretim işlemi kaynaklı veya ürün kaynaklı olabilir. Bilinen yapılarda, kısmi olarak karakterize edilmiş veya tanımlanmamış olabilirler.
- İşlem kaynaklı safsızlıklar: Üretim işleminden gelen safsızlıklardır.
- hücre substratları (konakçı hücre proteinleri, konakçı hücre DNA’sı)
- hücre kültürü bileşenleri (indükleyici ajanlar, antibiyotikler, besiyeri bileşenleri)
- saflaştırma kaynaklı (reçine, tampon maddesi)
- Ürün kaynaklı safsızlıklar: Üretim ve/veya saklama sırasında ortaya çıkan moleküler değişikliklerdir.
- bazı parçalanma ürünleri, prekürsörler
- yüksek ve düşük MA’lı safsızlıklar
- agregatlar, dimerler
- yük heterojenlikleri
- kesilmiş formlar
- glikozilasyon izoformu
Ürün ya da üretim işlemi kaynaklı safsızlıklar ve post translasyonel modifikasyonlar immünojenisiteye neden olabilir.
Kontaminantlar, ürün içerisinde tesadüf olarak bulunan, üretim işleminin bir parçası olmayan kimyasal ve biyokimyasal materyaller (mikrobiyal proteazlar) ve/veya mikrobiyal parçalardır. Etkin madde ve ürün özellikleri için uygun işlem içi kabul kriterleri ve aksiyon limitleriyle kontrol altına alınmalıdır.
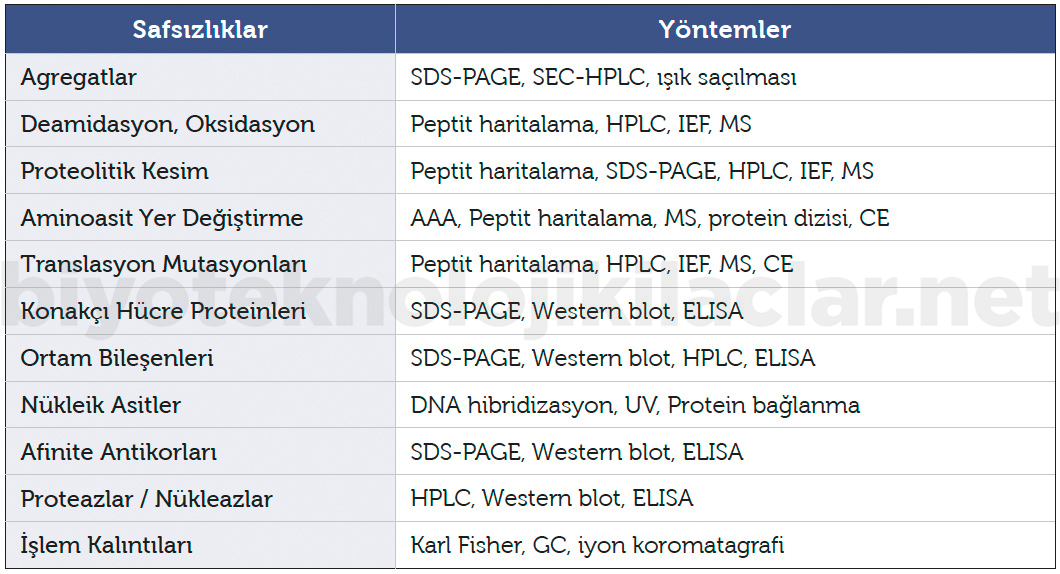
Üretim ve/veya saklama sırasında ortaya çıkan anlamlı miktarda parçalanma ürünleri belirli kabul kriterleri çerçevesinde test edilmelidir. Safsızlıklar farmakope ve kılavuzlar limitlerinde kabul edilebilir. Safsızlıkların tayininde kullanılan analitik yöntemler Tablo 2’de verilmiştir (16).
Tablo 2. Safsızlıkların tayininde kullanılan analitik yöntemler
e) Miktar tayini
Kantitasyon protein içeriği olarak ölçülür. Referans ilaçla karşılaştırılabilir güçte, aynı birimde ifade edilmeli, uygun fizikokimyasal yöntemlerle tayin edilmelidir.
Protein içeriği tayini için çeşitli protein tayin yöntemleri (Keldahl, Lowry, Bradford), absorbans ölçümü (280 nm), HPLC ve SDS-PAGE yöntemleri kullanılır (17).
3. Spesifikasyonlar
Karakterizasyonu yapılan etkin madde ve bitmiş ürüne ait görünüm, tanıma, saflık ve safsızlıklar, potens, miktar, sterilite, endotoksin, mikrobiyal limitler, liyofilize ürünler için nem içeriği gibi spesifikasyonların tanımlanması gerekmektedir. Bitmiş ürün spesifikasyonları için farmakope testleri kullanılır. Ürünün önerilen raf ömrü stabilite çalışmaları sonucunda belirlenir. Biyobenzer ilaç ile referans ilaç arasında karşılaştırmalı, gerçek zamanlı, gerçek koşullara dayalı stabilite çalışmaları gerekli değildir (1,2,11).
Biyoteknolojik ilaçların üretim süreçlerindeki klonlama vektörü, konakçı hücre, fermantasyon koşulları, saflaştırma işlemleri, formülasyon, primer ambalaj materyali gibi değişkenlerin ürünün kalitesi, etkililiği ve güvenliliği üzerine etkilerini saptamak için karşılaştırılabilirlik kavramından söz edilir. Biyobenzer ilaçların üretim süreçlerinin kendilerine özgü olması nedeniyle, referans ilacın kalite özellikleri ile tamamen aynı olması gerekli değildir. Post translasyonel modifikasyonlar nedeniyle etkin maddenin yapısında küçük farklılıklar olabilir. Biyobenzer ilaç ile referans ilacın kalite özellikleri arasındaki küçük farklar, kalite karşılaştırılabilirlik çalışmaları sürecinde analitik çalışmalar ile ortaya konabilir. Ayrıntılı karakterizasyon çalışmaları ile moleküler benzerlik, biyoaktivite ve safsızlıklar tayin edilmelidir. Gözlenen farklılıkların kabul edilebilir düzeyde olup olmadığı karşılaştırmalı in vitro ve in vivo klinik dışı çalışmalar ve klinik çalışmalar ile ortaya konarak, minör değişikliklerin etkililik ve güvenliliği değiştirmediği kanıtlanmalıdır.